Tıbbi Cihaz Tüzüğü (MDR 2017/745), AB genelinde regülatif süreci güçlendirmek için birçok yeni mekanizma getirmiştir. Belirli kategorilerdeki yüksek riskli cihazlar için bu mekanizmaların en önemlilerinden biri de Uzman Panel Klinik Değerlendirme Danışma Prosedürü'dür (CECP). Bu prosedür, cihazın uygunluk değerlendirmesi sırasında zorunlu olarak devreye girebilir.
CECP, yüksek riskli cihazlar için AB'deki en üst düzey bilimsel ve klinik incelemedir. Bu mekanizmanın kapsamını ve işleyişini doğru anlamak; üreticilerin sağlıklı proje planlaması, zaman çizelgelerini verimli yönetmesi ve sertifikasyon hedeflerine ulaşması için kritik önem taşımaktadır.
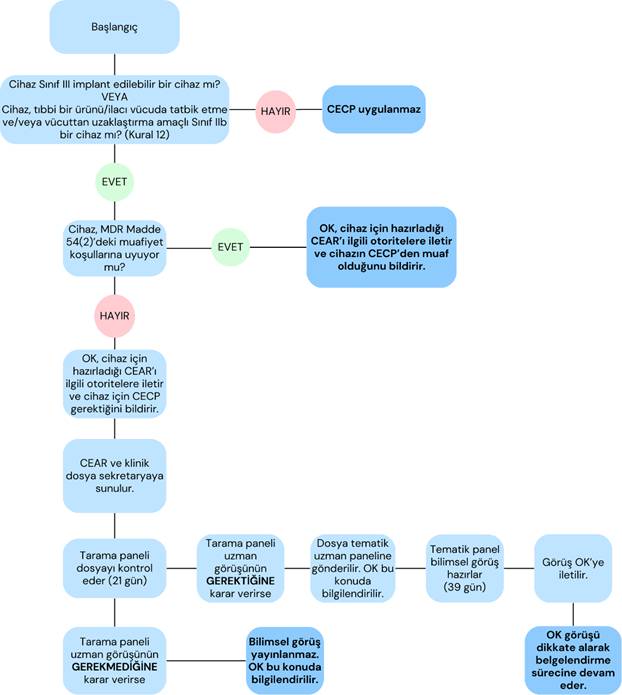
Hangi Cihazlar Uzman Panel Görüşü Gerektirir?
MDR Madde 54(1) uyarınca, uzman paneli görüşü iki cihaz kategorisi için zorunludur:
- Tüm Sınıf III implante edilebilir cihazlar
- Tıbbi bir ürünü/ilacı vücuda tatbik etme ve/veya vücuttan uzaklaştırma amaçlı tüm Sınıf IIb aktif cihazlar (Kural 12).
Bununla birlikte, MDR Madde 54(2)'de yer alan muafiyet koşullarına göre, CECP şu durumlarda gerekli değildir:
- MDR kapsamında daha önce düzenlenmiş sertifikaların yenilenmesi.
- Halihazırda piyasada bulunan bir cihazda, fayda–risk oranını olumsuz etkilemeyen değişiklikler (MDD veya MDR kapsamında).
- Miras (legacy) cihazlarda etiket güncellemesi gibi MDR’ye uyum amaçlı hukuki/idari değişiklikler için CECP aranmaz.
- Ancak yeni bir endikasyon eklenmesi, tasarım veya materyal değişikliği gibi majör veya klinik etkisi olan değişiklikler CECP gerekliliği doğurabilir.
- Yayınlanmış Ortak Spesifikasyonlara (CS) uyan cihazlar.
Uzman Panellerin Rolü ve Amacı
Uzman Paneller, Avrupa Komisyonu tarafından MDR Madde 106 uyarınca atanan bağımsız danışma gruplarıdır. Temel amaçları, yukarıdaki kriterleri karşılayan yüksek riskli tıbbi cihazların klinik değerlendirmesine ilişkin tutarlı ve üst düzey bilimsel görüşler sunmaktır.
Bu işlevin merkezileştirilmesiyle Avrupa Komisyonu, AB genelinde güvenlik ve performans standartlarını uyumlu hale getirmeyi hedeflemektedir. Uzman Paneller, Avrupa İlaç Ajansı'nın (EMA) bilimsel ve idari desteğiyle çalışmaktadır.
CECP Süreci Nasıl İşler?
MDR Madde 54(1) kapsamında CECP gerektiren cihazlar için Onaylanmış Kuruluş (OK), öncelikle Madde 54(2)’deki muafiyet koşullarının uygulanıp uygulanamayacağını değerlendirir. Değerlendirme sonucunu Yetkili Makamlara ve Komisyona bildirir. CECP gerekiyor ise OK, uzman panel başvurusunu başlatır.
Dosya tarama paneline ulaştığında, aşağıdaki üç kriter üzerinden bilimsel görüş gerekip gerekmediğine karar verilir:
- Cihazın veya ilgili klinik prosedürün yenilik düzeyi ve olası büyük klinik veya halk sağlığı etkisi.
- Bilimsel olarak geçerli sağlık endişeleri (örn. bileşenler, kaynak materyal veya cihaz arızasının etkisi) nedeniyle belirli bir cihaz kategorisi veya grubu için fayda-risk profilinde ciddi olumsuz değişiklik.
- Belirli bir cihaz kategorisi veya grubu için bildirilen ciddi olay oranında anlamlı bir artış (MDR Madde 87).
Tarama paneli tarafından bilimsel görüş gerektiğine karar verilirse, ilgili tematik uzman paneli dosyayı inceleyerek görüşünü yayınlar ve bu görüş OK ile paylaşılır. Görüş teknik olarak bağlayıcı değildir; ancak OK değerlendirmesinde, görüşü dikkate almak zorundadır. OK panelin tavsiyesini uygulamama kararı alırsa, bunu yazılı olarak gerekçelendirmesi gerekir.
Yayınlanmış Uzman Panel Görüşleri
Tüm Uzman Panel bilimsel görüşleri Avrupa Komisyonu tarafından yayınlanır. Günümüze kadar yayınlanmış olan görüşleri bu bağlantıdan inceleyebilirsiniz: Yayınlanmış Bilimsel Görüşler
Avrupa Komisyonu'nun CECP bildirimlerine ilişkin ikinci yıllık raporu (Temmuz 2022–Haziran 2023), sistemin pratikte nasıl işlediğini sayısal olarak göstermektedir:
- Toplam 353 bildirimin 317'si (%89,8) CECP’den muaf tutulan cihazlara aittir.
Bu muafiyetlerin %99,1'i, aynı üreticinin aynı kullanım amacıyla piyasada mevcut olan cihazlarda yaptığı, fayda-risk dengesini olumsuz etkilemeyen değişiklikler sebebiyledir.
- En sık muaf olarak bildirilen cihaz türleri:
- Ortopedik protezler (%30,0)
- İmplant edilebilir protez cihazlar (%13,9)
- Vasküler/kardiyak protezler (%12,0)
Teorik olarak bakıldığında, oldukça geniş bir cihaz portföyü için uzman paneli görüşü gerekebilir. Ancak yayınlanan veriler, özellikle güvenlik profili kanıtlanmış ve sınırlı değişiklikler yapılmış cihazlarda çoğu zaman bilimsel görüşe gerek duyulmadığını göstermektedir.
Uzman Panellerin Teknik Dokümantasyon Üzerindeki Etkisi
Uzman Panel sistemi, merkezi bir bilimsel inceleme mekanizması getirerek, yüksek riskli cihazlar için gerekli olan klinik kanıt düzeyini yükseltmektedir. Bu kapsamdaki üreticilerin, dokümantasyonlarının bu seviyede titiz bir değerlendirmeye hazır olduğundan emin olması gerekmektedir.
Hazırlık sürecinde özellikle şu noktalara dikkat edilmelidir:
- Güçlü Klinik Kanıt: Klinik Veri Değerlendirme Raporu (CER), tüm klinik kanıt boşluklarını kapsamlı bir şekilde ele almalıdır.
- Resmi Şablonların Kullanımı: Paneller standart formatlarla çalıştığı için MDCG şablonlarına uyum önemlidir.
- Açıklık ve Gerekçelendirme: Dokümantasyondaki belirsizlikler veya bilgi eksikleri, uzman panel görüşü gerekliliğini doğurabilir.
- Pre-Klinik Veriler: Paneller, klinik kanıtlarla birlikte pre-klinik ve üretim bilgilerini de değerlendirir.
Bu süreci başarıyla yönetmek, gelişen klinik standartları, güncel regülatif beklentileri ve her panelin bilimsel odağını yakından takip etmeyi gerektirir.
Size Nasıl Destek Olabiliriz?
CECP sürecini yönetmek, MDR kapsamındaki en zorlayıcı regülatif adımlardan biridir.
Desia'daki ekibimiz, yüksek riskli cihazlar için dokümantasyon hazırlama konusunda uzmanlaşmıştır. Hizmetlerimiz:
- Klinik Veri Değerlendirme Raporu (CER)
- PMCF Çalışma Planlaması
- Güvenlik ve Klinik Performans Özeti (SSCP)
- Biyouyumluluk Değerlendirmesi
- Kapsamlı Regülatif Danışmanlık
Desia bünyesinde, üreticilerin dokümantasyonlarındaki eksikleri tespit etmelerine, panel beklentilerine uygun bir hazırlık süreci yürütmelerine ve başvurularını eksiksiz şekilde tamamlamalarına destek oluyoruz.


